Asthma is a highly prevalent disorder associated with comorbidities, affecting over 350 million people worldwide and imposing a significant health and socioeconomic burden [1]. In 2015, as many as 400,000 asthma deaths have been reported [2].
Traditionally, asthma is diagnosed by (a history of) symptoms including wheeze, dyspnoea, chest tightness, and/or cough in combination with variable, often reversible, airway narrowing. Hallmarks of asthma underlying its clinical presentation include chronic airway inflammation, airway hyperresponsiveness, and structural changes within the airways referred to as ‘’airway remodelling’’. These characteristics are interrelated and have been shown to be more prominently present in uncontrolled disease [1]. Indeed, severe exacerbations reflecting an intense flair-up of the airway inflammation have been found to cause an accelerated decline in lung function in patients with more severe asthma [3,4]. This underscores the vital importance of reaching optimal asthma control and preventing exacerbations.
Despite increased awareness, patient counselling, implementation of comprehensive, evidence-based guidelines, and recent advances in pharmacological treatment options, data from studies and surveys demonstrate that the vast majority of asthma patients are inadequately controlled [5-7]. According to some studies, the percentage of uncontrolled asthmatic patients in Europe amounts up to 45% [8]. Apart from externally contributing factors such as non-adherence to treatments, inadequate inhalation technique, lifestyle, and environmental triggers, some asthma subtypes (especially severe asthma) may be truly difficult to control due to its heterogeneous nature and (relative) unresponsiveness to standard pharmacological treatment [9,10].
Patients with severe asthma, defined as those requiring treatment with high-dosed inhaled corticosteroids (ICS) plus a second controller and/or systemic corticosteroids (after other causes for lack of control, including adherence, inhalation technique, and comorbidities have been addressed), comprise approximately 5-10% of the entire asthma population but account for >80% of the total healthcare costs of asthma [9]. To aid the development of targeted treatment options, there is an urgent need to identify precipitating factors and inflammatory pathways of distinct subphenotypes of severe asthma.
From clinical, anatomical, pathophysiological, and inflammatory subsets to molecular pathways
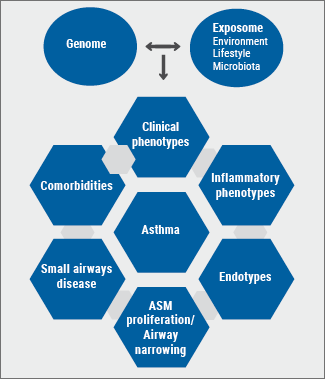
Based on growing insights into the underlying mechanisms of asthma during the past 3 decades, our look on asthma has evolved from the archetypic ‘’generalistic’’ syndrome to distinct disease subsets [10-12]. As in most diseases, the clinical expression (phenotype) of the different asthma subsets results from the combined effect of an individual’s genome and exposome (environmental exposures, microbiome, stress, lifestyle, antibiotics) and their interactions [13]. Consequently, asthma should in fact be evaluated and subsequently treated at several levels: i.e. clinical (age of onset, comorbidities, systemic presentations); anatomical (e.g. bronchiectasis, small airways disease/airway remodelling); physiological (e.g. airway narrowing, [non-]reversibility, airway hyperresponsiveness, airway trapping, ventilation, hypoxemia); and immuno-inflammatory (cells, cytokines, mediators, molecular components) (see Figure).
Figure: Diagram depicting multiple levels of (severe) asthma
Patients with adult-onset asthma have usually more severe disease and are less responsive to standard treatment with ICS compared to those with childhood-onset asthma [14]. Clinical predictors of persistence of adult-onset asthma include older age, comorbidities (especially nasal polyposis and NSAIDs-exacerbated respiratory disease [NERD)), more severe airway hyperresponsiveness, worse asthma control, and a need for more and higher doses of controller medications [15].
The involvement of small airways, characterised by distal lung inflammation and remodelling, is present in a large proportion of patients and is often a hidden cause of more severe asthma associated with a reduced response to standard therapy and a worse control [16-19]. Therefore, active identification (and subsequent treatment) of these 3-D anatomical site-specific phenotypes should be part of the assessment of severe (uncontrolled) asthma.
In the past two decades, non-invasive sampling methods combined with high-tech omics technologies helped to unravel the heterogeneity of severe asthma. Furthermore, unbiased cluster analyses of system biology-acquired data enabled to link distinct clinical phenotypes to underlying inflammatory and molecular pathways and to identify clinically applicable biomarkers [20,21].
Type 2 asthma
At the end of the last century, insights into the effects of a Western lifestyle on the immune system, complemented with animal data, led to the paradigm of asthma as mainly a T-helper type 2 (Th2)-driven disease with eosinophils as key players. In the late 1990s/beginning of 2000s, development and validation of non-invasive airway sampling techniques to assess airway inflammation (especially hypertonic saline induced sputum) [22] allowed identification of inflammatory subsets (phenotypes) in asthma: i.e. eosinophilic, neutrophilic, mixed, and pauci-granulocytic phenotypes [23]. Several "early" studies in (more) severe asthma pointed towards the clinical value to discriminate between non-eosinophilic asthma and eosinophilic asthma, the latter being more frequently associated with more severe exacerbations and accelerated lung function decline [24-26]. As eosinophilic inflammation is generally well-responding to corticosteroids, an RCT showed that guiding (ICS) treatment by levels of sputum eosinophils in moderate-to-severe asthma patients resulted in fewer exacerbations as compared to a standard (British Thoracic Society) treatment regimen based on clinical symptoms and signs [27].
More recently, in a study of patients with mild-to-moderate asthma, gene expression analyses provided insight into the underlying molecular pathways and revealed two distinct profiles, i.e. a Th2-high and a Th2-low profile based on the expression of IL-13-inducible genes. In contrast to Th2-low patients, those with a Th2-high profile responded well to ICS [28].
In the past decade, emerging evidence pointed towards a key role of the innate immune system in the pathophysiology of type 2 asthma [29]. The epithelial cytokines (so-called alarmins) IL-25, IL-33, and thymic stromal lymphopoietin (TSLP) are considered to initiate the type 2 immune response by activating the innate lymphoid cells (ILCs: ILC1, ILC2, ILC3). Similarly to their ‘’adaptive counterparts’’, the Th2 cells, activated ILC2s contribute to the immune response in asthma by the release of T(h)2 (now termed type 2 or T2) cytokines IL-4, IL-5, and IL-13 [30], thus linking innate and adaptive immunity [31]. Furthermore, both cell types can be activated by cysteinyl leukotrienes (CysLTs) and prostaglandin (PG)-D2 through interaction with the respective receptors (CysLT1R and DP2/CRTH2) on their cell surface [32,33].
Type 2 cytokines are key elements in the immunopathology of eosinophilic asthma which is present in approximately 50% of the asthma population [34]. IL-5 is involved in eosinophil maturation, release from the bone marrow and subsequent recruitment into the airway, while IL-4 and IL-13 are mainly involved in, respectively, IgE synthesis, eosinophil chemotaxis, mucus production, airway hyperresponsiveness, airway narrowing, and remodelling (subepithelial fibrosis, airway smooth muscle proliferation) [35].
Recent evidence from in vitro studies suggests that corticosteroid subsensitivity in ILC2 can be induced by exposure to TSLP, a dexamethasone-regulated IL-7R alpha ligand [36]. In line with this finding, increased numbers of ILC2s have been found in patients with more severe and often corticosteroid-insensitive or -resistant asthma [37,38]. These data support why type 2 asthma is often associated with corticosteroid subsensitivity, and therefore requires either treatment with oral corticosteroids or targeted therapies with monoclonal antibodies [39]. Additionally, recent evidence showed that IL-13 may desensitise beta2-receptors in human airways, which further underscores the urge of alternative treatments in type 2 asthma [40].
Non-type 2 asthma
It has been suggested that non-T2 or T2-low asthma is present in approximately 50% of (adult) patients, has usually a less severe nature, and consists of several different phenotypes and endotypes (a disease subtype defined by a distinct molecular mechanism). Triggers include infections, occupational irritants, and oxidative stress. The mechanisms of non-type 2 asthma are not well understood, but may comprise neutrophilic inflammation, Th17 pathway, neurogenic inflammation, barrier defects, airway smooth muscle abnormalities, etc. [35]. Therefore, these asthma subsets usually show a modest or no response to corticosteroids or T2-targeted therapies, while biomarkers and targeted treatment are presently unavailable and, hence, an unmet need.
Airway neutrophilia, often reflecting an underlying respiratory tract infection, can be treated with macrolides, while predominant airway hyperresponsiveness or airway narrowing without a clear inflammatory component can be addressed with bronchodilators or bronchial thermoplasty [41].
However, changes in clinical presentation within individual patients over time are consistent with observations that the underlying immune/inflammatory responses may vary over time and that different inflammatory phenotypes can co-exist in one patient [42]. Hence, regular re-evaluation of an individual’s disease status remains important.
Treatments targeting type 2 inflammation
Asthma management has gradually evolved from a ‘’one-size-fits-all’’ concept into a more personalised approach [1]. While the majority of patients with mild-to-moderate and uncomplicated asthma reach optimal control with ICS-containing therapy, a significant minority with more severe disease fails to reach (optimal) control with ICS as a result of the underlying immunopatho(physio)logy requiring a personalised approach with targeted therapy guided by biomarkers [39,43,44].
Interestingly, guidelines for severe asthma management do not yet insist on standard evaluation of the airway inflammatory components to guide treatment, although the nature of the airway inflammation clearly drives the choice of targeted therapy [1].
For type 2 asthma, several add-on treatments targeting different inflammatory pathways are currently available, while several new drugs are in different stages of clinical development (see Figure) [44]. For patients with a strong allergic component (history and serum IgE ≥30 IU/mL), apart from environmental or life style adjustments, add-on therapy with anti-IgE monoclonal antibodies is recommended for managing asthma (omalizumab is currently the only registered anti-IgE therapy), while for distinct patients with uncontrolled disease and house dust mite allergy, specific sublingual therapy may be considered [1].
Figure: Inflammatory pathways and biomarkers underlying type 2 asthma and targeted therapies (approved and in clinical development) [44]
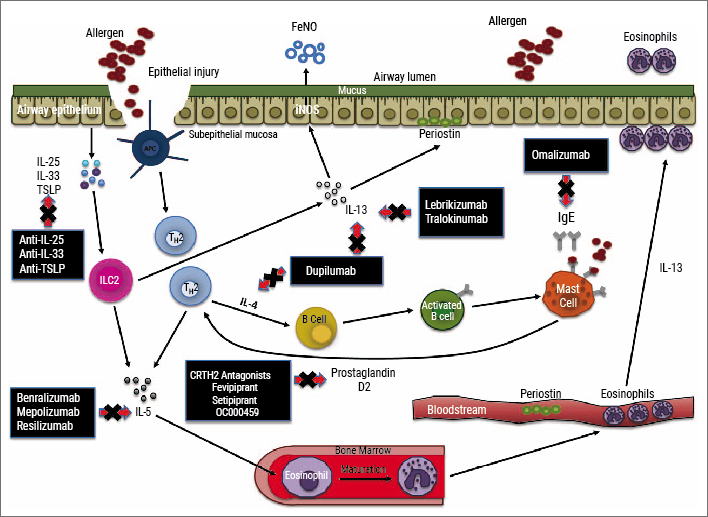
More recently, anti-IL-5 targeting monoclonal antibodies have been added to the stepwise approach for management of severe eosinophilic asthma [1]. To guide treatment, eosinophilia is preferably confirmed by induced sputum analysis (sputum eosinophils ≥3%) or by blood eosinophils as a surrogate marker. For blood eosinophils, different cut-offs have been applied across different studies and usually start at least ≥150 cells/µL (to over 300 cells/µL) [45]. Currently, there are three anti-IL-5 monoclonal antibodies available to treat severe adult asthma: mepolizumab, reslizumab, and (more recently) benralizumab. These agents have a slightly different target (IL-5 or its receptor), reflected by a different pharmacological profile, and offer a substantial reduction in asthma exacerbations and oral steroid intake independent of comorbid allergies [46].
Furthermore, various other monoclonal antibodies to treat asthma are currently under investigation. While targeting IL-13 proved less effective in phase 3 studies, blocking the joint IL-4/IL-13 pathway by targeting the IL-4R alpha with dupilumab showed promising effects on several health-related outcomes in type 2 diseases including atopic dermatitis, chronic rhinosinusitis with nasal polyps (CRSwNP), and severe asthma, irrespective of eosinophilia [47-49]. Fractionated exhaled nitric oxide (FeNO ≥25 ppb) is the best currently available biomarker to identify patients responding to IL-4/IL-13 targeted therapy [49]. Importantly, as systemic formulations, targeted therapies also showed beneficial effects on other aspects of asthma, such as comorbidities (e.g. CRSwNP, allergic rhinitis, atopic dermatitis) and may potentially also prove effective for small airways disease.
Future type 2 asthma treatments targeting alarmins (e.g. TSLP, IL-33), kinases (e.g. JAK, PI3K), and CRTH2/DP2 seem promising in distinct subsets and the results of phase 3 clinical trials are awaited [10].
Summary
- Chronic airway inflammation is a key feature of asthma and a risk factor of (severe) asthma exacerbations with subsequent accelerated lung function decline
- Asthma comprises abnormalities at several different levels (including comorbidities and small airways) which should be explored, evaluated, and adequately treated to achieve optimal control
- Severe asthma (5-10% of asthma population), is difficult to control and accounts for >80% of the total healthcare costs
- Severe asthma is a heterogeneous disease, roughly consisting of non-eosinophilic and eosinophilic inflammatory phenotypes – the latter being more frequently associated with more severe exacerbations and accelerated lung function decline
- Eosinophilic severe asthma can be either ‘’allergic’’ or ‘’non-allergic’’ – both being driven by type 2 inflammatory pathways including cytokines IL-5, IL-4, and IL-13.
- Although eosinophilic asthma usually responds well to treatment with inhaled corticosteroids, non-allergy-driven type 2 asthma (e.g. with chronic rhinosinusitis with nasal polyposis, NERD) is often corticosteroid-subsensitive.
- Several therapies targeting type 2 inflammation have been developed.
- Presently available biomarkers of type 2 inflammation comprise: sputum and blood eosinophils, FeNO, IgE, and periostin
- Non-type 2 asthma is less well-defined in terms of underlying mechanisms and biomarkers.
During the ERS 2018, long-term safety and efficacy data were presented from 2 ‘post-phase 3’ extension studies with approved anti-IL-5 biologics. The first study, COSMEX, evaluated the long-term safety and effectiveness of mepolizumab in an open label extension of the previous phase 3 COSMOS trials [50]. In this study, 339 patients with very severe, life-threatening, eosinophilic asthma received mepolizumab (100 mg every 4 weeks) for additionally up to 172 weeks. Overall, 93% of patients reported on-treatment adverse events (AEs), which occurred with a frequency of >10%, similar to the previous shorter-term placebo-controlled trials. The frequency of serious AEs was low. Effectiveness of mepolizumab treatment was sustained with substantial reductions in exacerbation rates and oral corticosteroids use, for up to 4.5 years.
The second study, BORA, reported on the long-term safety and effectiveness of benralizumab during an extension of the previous phase 3 trials SIROCCO and CALIMA [51]. In this study, 793 patients with uncontrolled severe eosinophilic asthma were enrolled and 512 continued benralizumab (30 mg every 4 weeks or every 8 weeks), while 281 continued placebo for another 52 weeks as in their previous study. In the BORA study, benralizumab treatment showed similar safety and tolerability outcomes to those in the previous phase 3 trials. Only very few serious AEs were reported, leading to discontinuation in 2-3% of patients. Long-term effectiveness was also similar and consistent with preceding studies, with approximately 75% of patients on benralizumab remaining exacerbation-free.
- Global Strategy for Asthma Management and Prevention. 2018 GINA Report.
- Soriano JB, et al. Lancet Respir Med. 2017;5(9):691-706.
- Bai TR, et al. Eur Respir J. 2007;30(3):452-6.
- O'Byrne PM, et al. Am J Respir Crit Care Med. 2009;179(1):19-24.
- Bateman ED, et al. Am JRespir Crit Care Med. 2004;170(8):836-44.
- Braido F, et al. Respir Res. 2016;17:51.
- Sastre J, et al. World Allergy Organ J. 2016;9(1):13.
- Price D, et al. NPJ Prim Care Respir Med. 2014;24:14009.
- Chung KF, et al. Eur Respir J. 2014;43(2):343-73.
- Israel E, Reddel HK. New Engl J Med. 2017;377(10):965-76.
- Diamant Z, et al. Respir Med. 2007;101(3):378-88.
- Wenzel SE. Nature Med. 2012;18:716.
- Von Mutius E. J Allergy Clin Immunol. 2009;123(1):3-11.
- Dunn RM, et al. Allergy. 2018;73(2):284-94.
- Westerhof GA, et al. J Allergy Clin Immunol. 2018;141(1):104-9.e3.
- Lipworth B, et al. Lancet Respir Med. 2014;2(6):497-506.
- Manoharan A, et al. Eur Respir J. 2014;44(5):1353-5.
- Weitoft M, et al. Respir Res. 2014;15(1):67.
- Wisniewski JA, et al. J Allergy Clin Immunol. 2018;141(6):2048-60.e13.
- Moore WC, et al. J Allergy Clin Immunol. 2014;133(6):1557-63.e5.
- Hekking P-P, et al. J Allergy Clin Immunol. 2018;141(4):1280-90.
- Gibson PG, et al. Agents Actions Suppl. 1990;30:161-72.
- Wenzel SE. Lancet. 2006;368(9537):804-13.
- Wenzel SE, et al. Am J Respir Crit Care Med. 1999;160(3):1001-8.
- Pavord ID, et al. Lancet. 1999;353(9171):2213-4.
- Ortega H, et al. J Allergy Clin Immunol In Pract. 2018;6(3):980-6.e1.
- Green RH, et al. Lancet. 2002;360(9347):1715-21.
- Woodruff PG, et al. Am J Respir Crit Care Med. 2009;180(5):388-95.
- Kortekaas Krohn I, et al. Allergy. 2018;73(4):837-50.
- Bartemes KR, et al. J Allergy Clin Immunol. 2014;134(3):671-8.e4.
- Lambrecht BN, Hammad H. Nature Immunol. 2015;16(1):45-56.
- Xue L, et al. J Allergy Clin Immunol. 2014;133(4):1184-94.
- Salimi M, et al. J Allergy Clin Immunol. 2017;140(4):1090-100.e11.
- Fahy JV. Nature Rev Immunol. 2015;15(1):57-65.
- Robinson D, et al. Clin Exp Allergy. 2017;47(2):161-75.
- Liu S, et al. J Allergy Clin Immunol. 2018;141(1):257-68.e6.
- Nagakumar P, et al. J Allergy Clin Immunol. 2016;137(2):624-6.e6.
- Smith SG, et al. J Allergy Clin Immunol. 2016;137(1):75-86.e8.
- Mukherjee M, et al. Curr Opin Pulm Med. 2017;23(1):78-88.
- Albano GD, et al. J Allergy Clin Immunol. 2015;135(5):1144-53.e1-9.
- Svenningsen S, Nair P. Front Med. 2017;4:158.
- Cosmi L, et al. J Allergy Clin Immunol. 2010;125(1):222-30.e1-4.
- Haldar P, et al. Am J Respir Crit Care Med. 2008;178(3):218-24.
- Parulekar AD, et al. Curr Opin Pulm Med. 2018;24(1):50-5.
- Yancey SW, et al. J Allergy Clin Immunol. 2017;140(6):1509-18.
- Farne HA, et al. Cochrane Database Syst Rev. 2017;9:Cd010834.
- Bachert C, et al. JAMA. 2016;315(5):469-79.
- Wang FP, et al. J Dermatol Sci. 2018;90(2):190-8.
- Castro M, et al. N Engl J Med. 2018;378(26):2486-96.
- Albers F, et al. OA3566; ERS 2018; Paris, France.
- Busse BW, et al. OA3567; ERS 2018; Paris, France.
Posted on
Table of Contents: ERS 2018
Featured articles
Letter from The Editor
[Long Read] Current Look on Asthma
COPD: Triple Therapy, MABA and Antibiotics
Landmark triple therapy trials
ICS: to use or not to use?
MABA, and novel LAMA
Macrolide antibiotics and trial with azithromycin
Current Look on Asthma
[Long Read] Current Look on Asthma
Endoscopic Solutions
Endoscopic treatment of emphysema
Endoscopic treatment of asthma
Endoscopic treatment of chronic bronchitis
PAH
Balloon pulmonary angioplasty for CTEPH
New therapeutic targets: moving form pre-clinical data to phase 2 studies
IPF
Gastroesophageal reflux, IPF and lessons learned
Oncology
ALK inhibition, guidelines, liquid biopsies, and immunotherapy
Brain metastases, lung cancer and interstitial lung disease
site created by:

© 2024 Medicom Medical Publishers. All rights reserved. Terms and Conditions | Privacy Policy
HEAD OFFICE
Laarderhoogtweg 25
1101 EB Amsterdam
The Netherlands
T: +31 85 4012 560
E: publishers@medicom-publishers.com