PsA pathogenesis
Psoriatic arthritis is an inflammatory arthritis that occurs in about a quarter of patients with cutaneous psoriasis and most often begins after the onset of skin disease. PsA is highly heritable, but a greater contribution to disease susceptibility is attributable to psoriasis-associated gene variants. Class I HLA B alleles are most strongly associated with PsA. Several environmental factors, particularly trauma, have been identified as potential triggers of PsA. Recent pathogenetic studies using samples from the synovial fluid, synovium, skin and enthesis indicate the importance of tissue resident memory cells as well as CD8 T cells in disease pathogenesis. γδT-cells play an important role in the enthesis. Anti-cytokine therapies also indicate tissue cytokine hierarchy with IL-23 and IL-17 being important for skin psoriasis, TNF, IL-17and IL-23 for peripheral synovitis, TNF and IL-17 for axial arthritis, IL-17 and IL-12/23 for enthesitis and TNF and IL-12/23 for inflammatory bowel disease. The pathogenesis of PsA is complex with an interplay between genetic and environmental factors leading to aberrant immune activation possibly in the skin, gut or enthesis leading to sustained inflammation in the synovium and periarticular structures, leading to bone loss as well as new-bone formation.
https://doi.org/10.55788/d1cace39
INTRODUCTION
Psoriatic Arthritis (PsA) is defined as an inflammatory arthritis associated with cutaneous psoriasis, and is classified according to the Classification of PsA criteria (CASPAR)[1]. PsA is a heterogeneous disease: manifestations include synovitis, enthesitis, dactylitis, axial arthritis in addition to skin and nail psoriasis. These manifestations may not be present in all patients and can vary with time[2]. There is also heterogeneity in joint damage progression as well as treatment response. PsA is present in about a quarter of patients with psoriasis[3]. Most patients develop PsA after the onset of skin psoriasis[2]. Thus, a model of studying pathogenesis of PsA is to study the mechanisms that lead to the onset and progression of inflammation at the musculoskeletal structures in patients with psoriasis[4]. In this model, some patients with psoriasis develop a phase of aberrant immune response in the skin, gut or enthesis that subsequently leads to subclinical inflammation where sensitive imaging methods demonstrate musculoskeletal inflammation but the patient is asymptomatic. This is followed by prodromal phase whereby patients experience symptoms of joint pain and fatigue with no overt signs of arthritis. Finally, patients develop signs and symptoms of overt PsA and may now be diagnosed with PsA and satisfy CASPAR[4].
GENETIC FACTORS
Genetic factors play a significant role in psoriatic disease susceptibility. Psoriasis and psoriatic arthritis are highly heritable[5]. In a study in the Icelandic population, the recurrence of risk ratio (λ) for PsA in the first-degree relatives was found to be 39.2[6]. This ratio declines rapidly when going down the degree of relatedness becoming non-significant with 5th degree relatives. Since the λ in the first-degree relatives was much hiher in patients with PsA than that for psoriasis alone, it was assumed that the genetic burden for PsA is higher than that for psoriasis. However, subsequent studies have not found many PsA-specific genes. Using data from genome-wide association studies (GWAS), Li et al demonstrated that both cutaneous psoriasis and PsA exhibit considerable heritability, but a greater contribution comes from cutaneous psoriasis[7]. Thus, there might not be many ‘PsA-specific’ gene variants. The SNPs located within the major histocompatibility region (MHC) on chromosome 6p explained a significant proportion of the heritability[7].There have been few GWAS that have compared patients with PsA to those with psoriasis without PsA (PsC). These studies have demonstrated that only variants on the MHC are significantly different between PsA and PsC at genome-wide significance[8]. A variant near IL23R and another near TNFAIP3 were more strongly associated with PsA than PsC[8]. Further interrogation of the MHC region indicated that the risk heterogeneity between PsA and PsC was driven by HLA-B amino acid position 45, where the presence of glutamine (instead of methionine, lysine or threonine) increased the risk for PsA (odds ratio: 1.46, p= 2.9 × 10−12)[9]. This association was stronger than individual HLA C and B alleles. Interestingly, HLA alleles associated with PsA including HLA-B*27, -B*38, -B*39, as well as a number of other alleles, carries glutamine at position 45[9]. In a similar study, Bowes et al showed that amino acid position 97 of HLA-B differentiates PsA from PsC[10]. There is a wide range in the time interval between the onset of psoriasis and PsA[11]. HLA alleles influence this relationship; patients with HLA-B*27 have a much shorter interval compared to those with HLA C*06[12].
ENVIRONMENTAL FACTORS
Complex genetic diseases such as PsA are likely triggered by environmental factors. Overt physical trauma (deep Koebner phenomenon), microtrauma, stress, infections (requiring antibiotics), smoking and drugs such as retinoids have been associated with PsA[13]. Obesity and metabolic syndrome and associated manifestations, uveitis, depression, and thyroid disease may also provide second hits. The mechanisms by which these lead to PsA is not known; the microbiome may play a significant role[14].
PATHOGENETIC INSIGHTS FROM TARGET TISSUESSYNOVIUM
There are no human studies evaluating immune activation prior to the onset of overt PsA. Recent studies have evaluated target tissues to describe immune activation in order to obtain clues to pathogenesis. Penkava et al studied the cellular landscape of blood, synovial fluid, and synovial tissue at single-cell resolution from a small number of PsA patients[15]. They identified significant expansions of synovial memory CD8 and memory CD4 T cells in all patients compared to blood. Plasmacytoid and conventional dendritic cells were also expanded in the synovial fluid. B cells and basophils were depleted, and monocytes, γδT, MAIT and NK cells were unchanged. When comparing gene expression between synovial fluid and blood T cells, an increased expression of activation and effector markers in synovial fluid in the HLA-DR-low CD8, HLA-DR- high CD8 and ZNF683+ CD8 clusters were observed. There was synovial clonal expansion within the ZNF683+ CD8 cluster indicating expansion of tissue resident memory T cells. Interestingly, CXCR3 was the most strongly expressed chemokine receptor gene in synovial-enriched T-cell clones. The CXCR3 ligands CXCL10, previously identified as a biomarker of progression from psoriasis to PsA, and CXCL9 were highly enriched in the synovial fluid compared to blood[15,16]. Clonal expansion of CD8 T cells indicate that arthritogenic antigen(s) may be driving immune response in the synovium. Another single cell sequencing study in PsA SF identified 12 different cell populations, with the most dominant being monocytes/macrophages. The monocytes/macrophages were comprised of four subpopulations, three of which were large representing classical, non-classical and intermediate cells. The classical monocytes/macrophages were reduced in PsA compared to other arthritides (osteoarthritis, rheumatoid arthritis), whilst the intermediate population was increased[17].Importantly, histopathological and gene expression studies have emphasized the significant histopathological and molecular heterogeneity of the synovitis in PsA. Nerviani et al performed gene expression analysis on 14 matched synovial tissue, lesional and adjacent non-lesional skin[18]. They showed that the synovium clusters away from the skin, with a partial overlapping of lesional and non-lesional skin. Principal component analyses showed that IL17A/F, IL23R and IL21 were the major contributors of variation in lesional skin, whereas in the synovium, genes related to ectopic lymphoid structure formation (CXCL13, CXCR5) and the IL-23 axis (IL23A, IL12B, IL23R) together strongly contributed to the variation. Synovial IL-23p40/p19 and IL-23R protein expression correlates with the histological inflammatory status. There was also a positive correlation between IL-23p40/IL-23p19/IL-23R-positive cells and synovitis scores, and lower IL-23 cytokines/receptor tissue-expression in the pauci-immune compared with macrophage-rich histological pathotypes[18].
SKIN
Mediators originating in the inflamed skin could trigger musculoskeletal inflammation. This theory is supported by a recent study that demonstrated increased circulatory skin derived tissue resident memory CCR10+ CD8+ T cells in the peripheral circulation of PsA patients compared to patients with psoriasis[19]. However, these cells were not enriched in the synovial fluid. CD8+CCR10+ T cells co-expressed DNAM-1. DNAM-1 is an activating receptor, and TIGIT is an inhibitory receptor on T cells. CD8+CCR10+ T cells were typically DNAM-1high but had less TIGIT co-expression in PsA. Interestingly, CD8+CCR10+ T cells produced significantly more IL-17A and IL-22 compared to bulk CD8+ T cells on ex vivo restimulation and may be initiating synovial inflammation[19].
ENTHESIS
The entheses are important target tissues and may be the initial site of inflammation in PsA and other spondyloarthritides[20]. However, it is difficult to obtain biopsies of inflamed enthesis to study pathogenesis. Therefore, studies have used cadaveric tissues as well as spinous processes entheseal soft tissue (EST) and peri-entheseal bone (PEB) obtained during elective orthopaedic procedures. Cuthbert et al showed that γδT-cells are present in the EST and PEB and adjacent haematopoietic bone marrow and in the soft tissue of the ligaments[21]. γδT-cells were also observed in inflammatory infiltrate in ruptured Achilles’ tissue, indicating their presence at the sites of injury. The Vδ1 subset of γδT-cells had a far greater proportion of cells with a naïve phenotype compared with the Vδ2 subset. Vδ1 subset from peri-entheseal bone contained a greater proportion of the tissue resident memory phenotype compared to those from blood. Following stimulation with a combination of anti-CD3/CD28, IL-17A, IL-17F and IL-22 transcripts was detected in Vδ1 and Vδ2 subsets. However, IL-23 stimulation had almost no effect in the Vδ1 subset but caused a marked increase in the Vδ2 subset. This study demonstrated that spinal entheseal Vδ1 and Vδ2 subsets are tissue resident cells with inducible IL-17A production and that the Vδ1 subset does so independently of IL-23R expression[21]. These studies indicate the possible role of tissue resident Vδ1 γδT-cells in the entheses. The production of IL-17 independent of IL-23R expression might be the reason behind the lack of efficacy of IL-23 inhibitors in axial spondyloarthritis.
INSIGHTS FROM CLINICAL TRIALS
Anti-cytokine therapies have also provided us with insights into the importance of cytokines that drive inflammation in the different of PsA. Evidence from randomised clinical trials indicate tissue cytokine hierarchy with IL-23 and IL-17 being important for skin psoriasis, TNF, IL-17and IL-23 for peripheral synovitis, TNF and IL-17 for axial arthritis, IL-17 and IL-12/23 for enthesitis and TNF and IL-12/23 for inflammatory bowel disease[22].
RELATIONSHIP BETWEEN SKIN AND MUSCULOSKELETAL INFLAMMATION
The intimate relationship between skin and musculoskeletal inflammation begets the question whether the relationship between inflammation at the two sites is successive (changes in the skin triggering musculoskeletal inflammation) or synchronous (a common trigger leading to skin and musculoskeletal inflammation). Both mechanisms may be operative (figure). 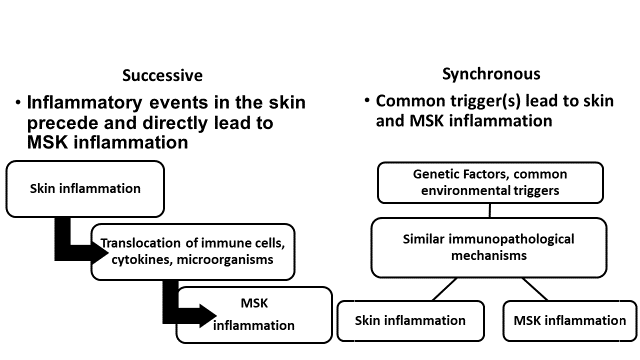
There are no human studies evaluating immune activation prior to the onset of overt PsA. Recent studies have evaluated target tissues to describe immune activation in order to obtain clues to pathogenesis. Penkava et al studied the cellular landscape of blood, synovial fluid, and synovial tissue at single-cell resolution from a small number of PsA patients[15]. They identified significant expansions of synovial memory CD8 and memory CD4 T cells in all patients compared to blood. Plasmacytoid and conventional dendritic cells were also expanded in the synovial fluid. B cells and basophils were depleted, and monocytes, γδT, MAIT and NK cells were unchanged. When comparing gene expression between synovial fluid and blood T cells, an increased expression of activation and effector markers in synovial fluid in the HLA-DR-low CD8, HLA-DR- high CD8 and ZNF683+ CD8 clusters were observed. There was synovial clonal expansion within the ZNF683+ CD8 cluster indicating expansion of tissue resident memory T cells. Interestingly, CXCR3 was the most strongly expressed chemokine receptor gene in synovial-enriched T-cell clones. The CXCR3 ligands CXCL10, previously identified as a biomarker of progression from psoriasis to PsA, and CXCL9 were highly enriched in the synovial fluid compared to blood[15,16]. Clonal expansion of CD8 T cells indicate that arthritogenic antigen(s) may be driving immune response in the synovium. Another single cell sequencing study in PsA SF identified 12 different cell populations, with the most dominant being monocytes/macrophages. The monocytes/macrophages were comprised of four subpopulations, three of which were large representing classical, non-classical and intermediate cells. The classical monocytes/macrophages were reduced in PsA compared to other arthritides (osteoarthritis, rheumatoid arthritis), whilst the intermediate population was increased[17].Importantly, histopathological and gene expression studies have emphasized the significant histopathological and molecular heterogeneity of the synovitis in PsA. Nerviani et al performed gene expression analysis on 14 matched synovial tissue, lesional and adjacent non-lesional skin[18]. They showed that the synovium clusters away from the skin, with a partial overlapping of lesional and non-lesional skin. Principal component analyses showed that IL17A/F, IL23R and IL21 were the major contributors of variation in lesional skin, whereas in the synovium, genes related to ectopic lymphoid structure formation (CXCL13, CXCR5) and the IL-23 axis (IL23A, IL12B, IL23R) together strongly contributed to the variation. Synovial IL-23p40/p19 and IL-23R protein expression correlates with the histological inflammatory status. There was also a positive correlation between IL-23p40/IL-23p19/IL-23R-positive cells and synovitis scores, and lower IL-23 cytokines/receptor tissue-expression in the pauci-immune compared with macrophage-rich histological pathotypes[18].
SKIN
Mediators originating in the inflamed skin could trigger musculoskeletal inflammation. This theory is supported by a recent study that demonstrated increased circulatory skin derived tissue resident memory CCR10+ CD8+ T cells in the peripheral circulation of PsA patients compared to patients with psoriasis[19]. However, these cells were not enriched in the synovial fluid. CD8+CCR10+ T cells co-expressed DNAM-1. DNAM-1 is an activating receptor, and TIGIT is an inhibitory receptor on T cells. CD8+CCR10+ T cells were typically DNAM-1high but had less TIGIT co-expression in PsA. Interestingly, CD8+CCR10+ T cells produced significantly more IL-17A and IL-22 compared to bulk CD8+ T cells on ex vivo restimulation and may be initiating synovial inflammation[19].
ENTHESIS
The entheses are important target tissues and may be the initial site of inflammation in PsA and other spondyloarthritides[20]. However, it is difficult to obtain biopsies of inflamed enthesis to study pathogenesis. Therefore, studies have used cadaveric tissues as well as spinous processes entheseal soft tissue (EST) and peri-entheseal bone (PEB) obtained during elective orthopaedic procedures. Cuthbert et al showed that γδT-cells are present in the EST and PEB and adjacent haematopoietic bone marrow and in the soft tissue of the ligaments[21]. γδT-cells were also observed in inflammatory infiltrate in ruptured Achilles’ tissue, indicating their presence at the sites of injury. The Vδ1 subset of γδT-cells had a far greater proportion of cells with a naïve phenotype compared with the Vδ2 subset. Vδ1 subset from peri-entheseal bone contained a greater proportion of the tissue resident memory phenotype compared to those from blood. Following stimulation with a combination of anti-CD3/CD28, IL-17A, IL-17F and IL-22 transcripts was detected in Vδ1 and Vδ2 subsets. However, IL-23 stimulation had almost no effect in the Vδ1 subset but caused a marked increase in the Vδ2 subset. This study demonstrated that spinal entheseal Vδ1 and Vδ2 subsets are tissue resident cells with inducible IL-17A production and that the Vδ1 subset does so independently of IL-23R expression[21]. These studies indicate the possible role of tissue resident Vδ1 γδT-cells in the entheses. The production of IL-17 independent of IL-23R expression might be the reason behind the lack of efficacy of IL-23 inhibitors in axial spondyloarthritis.
INSIGHTS FROM CLINICAL TRIALS
Anti-cytokine therapies have also provided us with insights into the importance of cytokines that drive inflammation in the different of PsA. Evidence from randomised clinical trials indicate tissue cytokine hierarchy with IL-23 and IL-17 being important for skin psoriasis, TNF, IL-17and IL-23 for peripheral synovitis, TNF and IL-17 for axial arthritis, IL-17 and IL-12/23 for enthesitis and TNF and IL-12/23 for inflammatory bowel disease[22].
RELATIONSHIP BETWEEN SKIN AND MUSCULOSKELETAL INFLAMMATION
The intimate relationship between skin and musculoskeletal inflammation begets the question whether the relationship between inflammation at the two sites is successive (changes in the skin triggering musculoskeletal inflammation) or synchronous (a common trigger leading to skin and musculoskeletal inflammation). Both mechanisms may be operative (figure).
The entheses are important target tissues and may be the initial site of inflammation in PsA and other spondyloarthritides[20]. However, it is difficult to obtain biopsies of inflamed enthesis to study pathogenesis. Therefore, studies have used cadaveric tissues as well as spinous processes entheseal soft tissue (EST) and peri-entheseal bone (PEB) obtained during elective orthopaedic procedures. Cuthbert et al showed that γδT-cells are present in the EST and PEB and adjacent haematopoietic bone marrow and in the soft tissue of the ligaments[21]. γδT-cells were also observed in inflammatory infiltrate in ruptured Achilles’ tissue, indicating their presence at the sites of injury. The Vδ1 subset of γδT-cells had a far greater proportion of cells with a naïve phenotype compared with the Vδ2 subset. Vδ1 subset from peri-entheseal bone contained a greater proportion of the tissue resident memory phenotype compared to those from blood. Following stimulation with a combination of anti-CD3/CD28, IL-17A, IL-17F and IL-22 transcripts was detected in Vδ1 and Vδ2 subsets. However, IL-23 stimulation had almost no effect in the Vδ1 subset but caused a marked increase in the Vδ2 subset. This study demonstrated that spinal entheseal Vδ1 and Vδ2 subsets are tissue resident cells with inducible IL-17A production and that the Vδ1 subset does so independently of IL-23R expression[21]. These studies indicate the possible role of tissue resident Vδ1 γδT-cells in the entheses. The production of IL-17 independent of IL-23R expression might be the reason behind the lack of efficacy of IL-23 inhibitors in axial spondyloarthritis.
INSIGHTS FROM CLINICAL TRIALS
Anti-cytokine therapies have also provided us with insights into the importance of cytokines that drive inflammation in the different of PsA. Evidence from randomised clinical trials indicate tissue cytokine hierarchy with IL-23 and IL-17 being important for skin psoriasis, TNF, IL-17and IL-23 for peripheral synovitis, TNF and IL-17 for axial arthritis, IL-17 and IL-12/23 for enthesitis and TNF and IL-12/23 for inflammatory bowel disease[22].
RELATIONSHIP BETWEEN SKIN AND MUSCULOSKELETAL INFLAMMATION
The intimate relationship between skin and musculoskeletal inflammation begets the question whether the relationship between inflammation at the two sites is successive (changes in the skin triggering musculoskeletal inflammation) or synchronous (a common trigger leading to skin and musculoskeletal inflammation). Both mechanisms may be operative (figure).
The intimate relationship between skin and musculoskeletal inflammation begets the question whether the relationship between inflammation at the two sites is successive (changes in the skin triggering musculoskeletal inflammation) or synchronous (a common trigger leading to skin and musculoskeletal inflammation). Both mechanisms may be operative (figure).
Figure. The relationship between skin and musculoskeletal inflammation in psoriatic arthritis. MSK- musculoskeletal.
SUMMARY AND CONCLUSION
Thus, the pathogenesis of PsA is complex with an interplay between genetic and environmental factors leading to aberrant immune activation possibly in the skin, gut or enthesis leading to sustained inflammation in the synovium and periarticular structures, leading to bone loss as well as new-bone formation. Taking into account the disease phenotypes and genetic (HLA) associations, Jadon et al proposed model of pathobiology of psoriatic disease[23]. They propose that amplification of the IL-23–IL-17 axis is initiated by activation of innate cells in the skin, entheses and gastrointestinal tract, ultimately resulting in the expansion of CD4+ and CD8+ Th1 and Th17 cells, which are expanded by IL-23 and IL-12 and produce TNF and IL-17. Different HLA alleles and/or haplotypes, T cell subsets and treatment response profiles are associated with different phenotypes. Synovial-predominant disease is associated with HLA-B*08:01:01, HLA-C*07:01:01, CD8+ engagement with Th1 cells and is responsive to TNF inhibition. Cutaneous-predominant disease is associated with HLA-B*57:01 and HLA-C*06:02, Th1 cell-driven and is responsive to IL-17 and IL-23 inhibition. Entheseal-predominant with or without axial disease is associated with the HLA-B*27:05:02 allele, involves engagement of both Th1 and Th17 cells that produce both TNF and IL-17, and is responsive to TNF and IL-17 inhibition. Arthritis mutilans likely represents a combination of these host genetic factors and T cell interactions[23].
ACKNOWLEDGMENTS
The author is supported by a Pfizer Chair Research Award, Rheumatology, University of Toronto.
REFERENCES
[1] W. Taylor, D. Gladman, P. Helliwell, A. Marchesoni, P. Mease, H. Mielants; CASPAR Study Group, “Classification criteria for psoriatic arthritis: development of new criteria from a large international study”, Arthritis Rheum, 2006 Aug;54(8):2665-73. doi: 10.1002/art.21972.
[2] C.T. Ritchlin, R.A. Colbert, D.D Gladman, “Psoriatic Arthritis’, N Engl J Med, 2017 Mar 9;376(10):957-970. doi: 10.1056/NEJMra1505557.
[3] F. Alinaghi, M. Calov, L.E. Kristensen, D.D. Gladman, L.C. Coates, D. Jullien, et al, “Prevalence of psoriatic arthritis in patients with psoriasis: A systematic review and meta-analysis of observational and clinical studies”, J Am Acad Dermatol. 2019 Jan;80(1):251-265.e19. doi: 10.1016/j.jaad.2018.06.027.
[4] J.U. Scher, A. Ogdie, J.F. Merola, C. Ritchlin, “Preventing psoriatic arthritis: focusing on patients with psoriasis at increased risk of transition”, Nat Rev Rheumatol. 2019 Mar;15(3):153-166. doi: 10.1038/s41584-019-0175-0.
[5] V. Chandran, C.T. Schentag, J.E Brockbank, F.J. Pellett, S. Shanmugarajah, S.M. Toloza et al, “Familial aggregation of psoriatic arthritis”, Ann Rheum Dis. 2009 May;68(5):664-7. doi: 10.1136/ard.2008.089367.
[6] A. Karason, T.J. Love, B. Gudbjornsson. “A strong heritability of psoriatic arthritis over four generations--the Reykjavik Psoriatic Arthritis Study”, Rheumatology (Oxford). 2009 Nov;48(11):1424-8. doi: 10.1093/rheumatology/kep243.
[7] Q. Li, V. Chandran, L. Tsoi, D. O'Rielly, R.P Nair, D. Gladman, et al, “Quantifying Differences in Heritability among Psoriatic Arthritis (PsA), Cutaneous Psoriasis (PsC) and Psoriasis vulgaris (PsV)”, Sci Rep. 2020 Mar 18;10(1):4925. doi: 10.1038/s41598-020-61981-5.
[8] P.E. Stuart, R.P. Nair, L.C. Tsoi, T. Tejasvi, S. Das, H.M. Kang et al, “Genome-wide Association Analysis of Psoriatic Arthritis and Cutaneous Psoriasis Reveals Differences in Their Genetic Architecture”, Am J Hum Genet. 2015 Dec 3;97(6):816-36. doi: 10.1016/j.ajhg.2015.10.019.
[9] Y. Okada, B. Han, L.C. Tsoi, P.E Stuart, E. Ellinghaus, T. Tejasvi et al, “Fine mapping major histocompatibility complex associations in psoriasis and its clinical subtypes”, Am J Hum Genet. 2014 Aug 7;95(2):162-72. doi: 10.1016/j.ajhg.2014.07.002.
[10] J. Bowes, J. Ashcroft, N. Dand, F. Jalali-Najafabadi, E. Bellou, P. Ho, et al. Cross-phenotype association mapping of the MHC identifies genetic variants that differentiate psoriatic arthritis from psoriasis. Ann Rheum Dis. 2017 Oct;76(10):1774-1779. doi: 10.1136/annrheumdis-2017-211414.
[11] W. Tillett, R. Charlton, A. Nightingale, J. Snowball, A. Green, C. Smith C, et al, “Interval between onset of psoriasis and psoriatic arthritis comparing the UK Clinical Practice Research Datalink with a hospital-based cohort”, Rheumatology (Oxford). 2017 Dec 1;56(12):2109-2113. doi: 10.1093/rheumatology/kex323.
[12] R. Winchester, G. Minevich, V. Steshenko, B. Kirby, D. Kane, D.A. Greenberg et al, “HLA associations reveal genetic heterogeneity in psoriatic arthritis and in the psoriasis phenotype”, Arthritis Rheum. 2012 Apr;64(4):1134-44. doi: 10.1002/art.33415.
[13] V. Chandran, S.P. Raychaudhuri, “Geoepidemiology and environmental factors of psoriasis and psoriatic arthritis”, J Autoimmun. 2010 May;34(3):J314-21. doi: 10.1016/j.jaut.2009.12.001.
[14] A.L. Carvalho, C.M. Hedrich, “The Molecular Pathophysiology of Psoriatic Arthritis-The Complex Interplay Between Genetic Predisposition, Epigenetics Factors, and the Microbiome”, Front Mol Biosci. 2021 Apr 1;8:662047. doi: 10.3389/fmolb.2021.662047.
[15] F. Penkava, M.D.C. Velasco-Herrera, M.D. Young, N. Yager, L.N. Nwosu, A.G. Pratt et al, “Single-cell sequencing reveals clonal expansions of pro-inflammatory synovial CD8 T cells expressing tissue-homing receptors in psoriatic arthritis”, Nat Commun. 2020 Sep 21;11(1):4767. doi: 10.1038/s41467-020-18513-6.
[16] F. Abji, K.A. Lee, R.A. Pollock, R. Machhar, R.J. Cook, V. Chandran, “Declining levels of serum chemokine (C-X-C motif) ligand 10 over time are associated with new onset of psoriatic arthritis in patients with psoriasis: a new biomarker?”, Br J Dermatol. 2020 Nov;183(5):920-927. doi: 10.1111/bjd.18940.
[17] F. Abji, M. Rasti, A. Gómez-Aristizábal, C. Muytjens, M. Saifeddine, K. Mihara et al, “Proteinase-Mediated Macrophage Signaling in Psoriatic Arthritis”, Front Immunol. 2021 Mar 8;11:629726. doi: 10.3389/fimmu.2020.629726.
[18] A. Nerviani, M.A. Boutet, W.S.G. Tan, K. Goldmann, N. Purkayastha, T.A. Lajtos et al, “IL-23 skin and joint profiling in psoriatic arthritis: novel perspectives in understanding clinical responses to IL-23 inhibitors”, Ann Rheum Dis. 2021 May;80(5):591-597. doi: 10.1136/annrheumdis-2020-218186.
[19] E.F. Leijten, T.S. van Kempen, M.A Olde Nordkamp, J.N. Pouw, N.J. Kleinrensink, N.L. Vincken, et al, “Tissue-Resident Memory CD8+ T Cells From Skin Differentiate Psoriatic Arthritis From Psoriasis”, Arthritis Rheumatol. 2021 Jul;73(7):1220-1232. doi: 10.1002/art.41652.
[20] S.Z. Aydin, C. Bridgewood, A. Zabotti, N. Girolimetto, D. McGonagle, “The transition from enthesis physiological responses in health to aberrant responses that underpin spondyloarthritis mechanisms”, Curr Opin Rheumatol. 2021 Jan;33(1):64-73. doi: 10.1097/BOR.0000000000000768.
[21] R.J. Cuthbert, A. Watad, E.M. Fragkakis, R. Dunsmuir, P. Loughenbury, A. Khan, et al, “Evidence that tissue resident human enthesis γδT-cells can produce IL-17A independently of IL-23R transcript expression”, Ann Rheum Dis. 2019 Nov;78(11):1559-1565. doi: 10.1136/annrheumdis-2019-215210.
[22] S. Siebert, N.L. Millar, I.B. McInnes, “Why did IL-23p19 inhibition fail in AS: a tale of tissues, trials or translation?”, Ann Rheum Dis. 2019 Aug;78(8):1015-1018. doi: 10.1136/annrheumdis-2018-213654.
[23] D.R. Jadon, C. Stober, S.R. Pennington, O FitzGerald, “Applying precision medicine to unmet clinical needs in psoriatic disease”, Nat Rev Rheumatol. 2020 Nov;16(11):609-627. doi: 10.1038/s41584-020-00507-9.
From: Proceedings of the 6th IFPA WPPACTable of Contents
©2024 the author(s). Published with license by Medicom Medical Publishers.
This an Open Access article distributed under the terms of the Creative Commons attribution-non Commercial license (http://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Posted on
site created by:

© 2024 Medicom Medical Publishers. All rights reserved.
Terms and Conditions
| Privacy Policy
The author is supported by a Pfizer Chair Research Award, Rheumatology, University of Toronto.
REFERENCES
[1] W. Taylor, D. Gladman, P. Helliwell, A. Marchesoni, P. Mease, H. Mielants; CASPAR Study Group, “Classification criteria for psoriatic arthritis: development of new criteria from a large international study”, Arthritis Rheum, 2006 Aug;54(8):2665-73. doi: 10.1002/art.21972.
[2] C.T. Ritchlin, R.A. Colbert, D.D Gladman, “Psoriatic Arthritis’, N Engl J Med, 2017 Mar 9;376(10):957-970. doi: 10.1056/NEJMra1505557.
[3] F. Alinaghi, M. Calov, L.E. Kristensen, D.D. Gladman, L.C. Coates, D. Jullien, et al, “Prevalence of psoriatic arthritis in patients with psoriasis: A systematic review and meta-analysis of observational and clinical studies”, J Am Acad Dermatol. 2019 Jan;80(1):251-265.e19. doi: 10.1016/j.jaad.2018.06.027.
[4] J.U. Scher, A. Ogdie, J.F. Merola, C. Ritchlin, “Preventing psoriatic arthritis: focusing on patients with psoriasis at increased risk of transition”, Nat Rev Rheumatol. 2019 Mar;15(3):153-166. doi: 10.1038/s41584-019-0175-0.
[5] V. Chandran, C.T. Schentag, J.E Brockbank, F.J. Pellett, S. Shanmugarajah, S.M. Toloza et al, “Familial aggregation of psoriatic arthritis”, Ann Rheum Dis. 2009 May;68(5):664-7. doi: 10.1136/ard.2008.089367.
[6] A. Karason, T.J. Love, B. Gudbjornsson. “A strong heritability of psoriatic arthritis over four generations--the Reykjavik Psoriatic Arthritis Study”, Rheumatology (Oxford). 2009 Nov;48(11):1424-8. doi: 10.1093/rheumatology/kep243.
[7] Q. Li, V. Chandran, L. Tsoi, D. O'Rielly, R.P Nair, D. Gladman, et al, “Quantifying Differences in Heritability among Psoriatic Arthritis (PsA), Cutaneous Psoriasis (PsC) and Psoriasis vulgaris (PsV)”, Sci Rep. 2020 Mar 18;10(1):4925. doi: 10.1038/s41598-020-61981-5.
[8] P.E. Stuart, R.P. Nair, L.C. Tsoi, T. Tejasvi, S. Das, H.M. Kang et al, “Genome-wide Association Analysis of Psoriatic Arthritis and Cutaneous Psoriasis Reveals Differences in Their Genetic Architecture”, Am J Hum Genet. 2015 Dec 3;97(6):816-36. doi: 10.1016/j.ajhg.2015.10.019.
[9] Y. Okada, B. Han, L.C. Tsoi, P.E Stuart, E. Ellinghaus, T. Tejasvi et al, “Fine mapping major histocompatibility complex associations in psoriasis and its clinical subtypes”, Am J Hum Genet. 2014 Aug 7;95(2):162-72. doi: 10.1016/j.ajhg.2014.07.002.
[10] J. Bowes, J. Ashcroft, N. Dand, F. Jalali-Najafabadi, E. Bellou, P. Ho, et al. Cross-phenotype association mapping of the MHC identifies genetic variants that differentiate psoriatic arthritis from psoriasis. Ann Rheum Dis. 2017 Oct;76(10):1774-1779. doi: 10.1136/annrheumdis-2017-211414.
[11] W. Tillett, R. Charlton, A. Nightingale, J. Snowball, A. Green, C. Smith C, et al, “Interval between onset of psoriasis and psoriatic arthritis comparing the UK Clinical Practice Research Datalink with a hospital-based cohort”, Rheumatology (Oxford). 2017 Dec 1;56(12):2109-2113. doi: 10.1093/rheumatology/kex323.
[12] R. Winchester, G. Minevich, V. Steshenko, B. Kirby, D. Kane, D.A. Greenberg et al, “HLA associations reveal genetic heterogeneity in psoriatic arthritis and in the psoriasis phenotype”, Arthritis Rheum. 2012 Apr;64(4):1134-44. doi: 10.1002/art.33415.
[13] V. Chandran, S.P. Raychaudhuri, “Geoepidemiology and environmental factors of psoriasis and psoriatic arthritis”, J Autoimmun. 2010 May;34(3):J314-21. doi: 10.1016/j.jaut.2009.12.001.
[14] A.L. Carvalho, C.M. Hedrich, “The Molecular Pathophysiology of Psoriatic Arthritis-The Complex Interplay Between Genetic Predisposition, Epigenetics Factors, and the Microbiome”, Front Mol Biosci. 2021 Apr 1;8:662047. doi: 10.3389/fmolb.2021.662047.
[15] F. Penkava, M.D.C. Velasco-Herrera, M.D. Young, N. Yager, L.N. Nwosu, A.G. Pratt et al, “Single-cell sequencing reveals clonal expansions of pro-inflammatory synovial CD8 T cells expressing tissue-homing receptors in psoriatic arthritis”, Nat Commun. 2020 Sep 21;11(1):4767. doi: 10.1038/s41467-020-18513-6.
[16] F. Abji, K.A. Lee, R.A. Pollock, R. Machhar, R.J. Cook, V. Chandran, “Declining levels of serum chemokine (C-X-C motif) ligand 10 over time are associated with new onset of psoriatic arthritis in patients with psoriasis: a new biomarker?”, Br J Dermatol. 2020 Nov;183(5):920-927. doi: 10.1111/bjd.18940.
[17] F. Abji, M. Rasti, A. Gómez-Aristizábal, C. Muytjens, M. Saifeddine, K. Mihara et al, “Proteinase-Mediated Macrophage Signaling in Psoriatic Arthritis”, Front Immunol. 2021 Mar 8;11:629726. doi: 10.3389/fimmu.2020.629726.
[18] A. Nerviani, M.A. Boutet, W.S.G. Tan, K. Goldmann, N. Purkayastha, T.A. Lajtos et al, “IL-23 skin and joint profiling in psoriatic arthritis: novel perspectives in understanding clinical responses to IL-23 inhibitors”, Ann Rheum Dis. 2021 May;80(5):591-597. doi: 10.1136/annrheumdis-2020-218186.
[19] E.F. Leijten, T.S. van Kempen, M.A Olde Nordkamp, J.N. Pouw, N.J. Kleinrensink, N.L. Vincken, et al, “Tissue-Resident Memory CD8+ T Cells From Skin Differentiate Psoriatic Arthritis From Psoriasis”, Arthritis Rheumatol. 2021 Jul;73(7):1220-1232. doi: 10.1002/art.41652.
[20] S.Z. Aydin, C. Bridgewood, A. Zabotti, N. Girolimetto, D. McGonagle, “The transition from enthesis physiological responses in health to aberrant responses that underpin spondyloarthritis mechanisms”, Curr Opin Rheumatol. 2021 Jan;33(1):64-73. doi: 10.1097/BOR.0000000000000768.
[21] R.J. Cuthbert, A. Watad, E.M. Fragkakis, R. Dunsmuir, P. Loughenbury, A. Khan, et al, “Evidence that tissue resident human enthesis γδT-cells can produce IL-17A independently of IL-23R transcript expression”, Ann Rheum Dis. 2019 Nov;78(11):1559-1565. doi: 10.1136/annrheumdis-2019-215210.
[22] S. Siebert, N.L. Millar, I.B. McInnes, “Why did IL-23p19 inhibition fail in AS: a tale of tissues, trials or translation?”, Ann Rheum Dis. 2019 Aug;78(8):1015-1018. doi: 10.1136/annrheumdis-2018-213654.
[23] D.R. Jadon, C. Stober, S.R. Pennington, O FitzGerald, “Applying precision medicine to unmet clinical needs in psoriatic disease”, Nat Rev Rheumatol. 2020 Nov;16(11):609-627. doi: 10.1038/s41584-020-00507-9.
From: Proceedings of the 6th IFPA WPPACTable of Contents
©2024 the author(s). Published with license by Medicom Medical Publishers.
This an Open Access article distributed under the terms of the Creative Commons attribution-non Commercial license (http://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Posted on
site created by:
© 2024 Medicom Medical Publishers. All rights reserved.
Terms and Conditions
| Privacy Policy
© 2024 Medicom Medical Publishers. All rights reserved. Terms and Conditions | Privacy Policy